- Have any questions? Contact us!
- info@dr-rath-foundation.org


Lipoprotein(a) Reduction by Ascorbate
October 4, 2017
A New Era in Medicine
October 4, 2017Reducing the Risk for Cardiovascular Disease with Nutritional Supplements
Matthias Rath M.D. (1992)
Journal of Orthomolecular Medicine, 7:153-162
Introduction
Cardiovascular Disease (CVD) is the most frequent cause of death in the industrialized world. In a series of recent papers I have contributed to an improved understanding about the pathogenesis of human CVD. It was shown that ascorbate deficiency is an important underlying factor and that all mechanisms known today leading to CVD can be triggered by ascorbate deficiency. This remarkable fact reflects the strong pressure during the evolution of man after loss of endogenous ascorbate synthesis. This pressure favored genetic and metabolic features contributing to avoidance of the fatal consequences of ascorbate deficiency and scurvy. The different mechanisms of human CVD known today therefore all compensate for impaired integrity and stability of the vascular wall caused by chronically low dietary ascorbate intake. If these mechanisms overshoot, heart attack, stroke and other forms of CVD develop. 1,2
In the first part of this paper I will marshal the evidence for the most frequent of these pathomechanisms. I will focus here on mechanisms related to lipid and lipoprotein deposition in the vascular wall and re-evaluate existing hypotheses. This re-evaluation is particularly necessary since cholesterol lowering concepts have become dominant factors in the public health debate. I will show that the most important among these overshooting defense mechanisms is the extracellular deposition of lipoprotein(a) in the vascular wall. On the basis of an improved understanding of these pathomechanisms I will present new therapeutic approaches including the reversibility of existing atherosclerotic deposits. Finally I will marshal the evidence for the particular value of nutritional supplements to achieve this therapeutic aim.
Lipoprotein(a), not LDL, is the Primary Risk Factor for CVD in Plasma
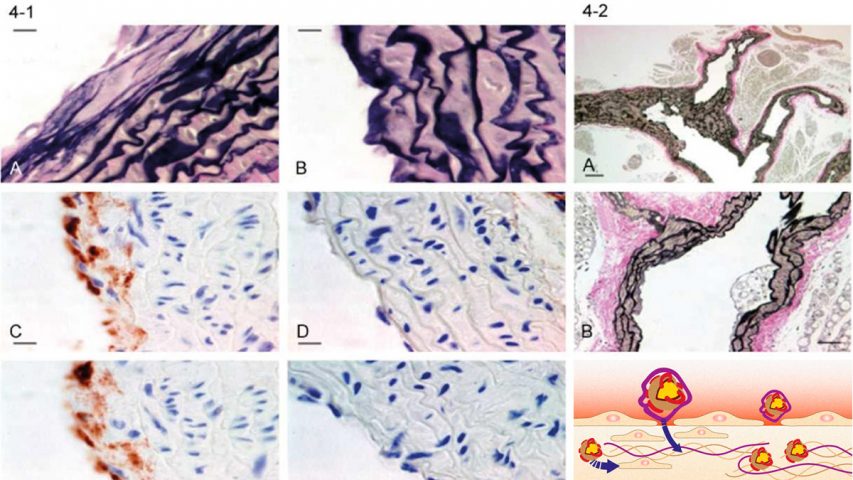
Present theories of human CVD are based on the concept that low-density lipoprotein (LDL) or LDL-cholesterol is the primary risk factor for CVD in plasma.3,4 A closer look at the available epidemiological data challenges this assumption. Lipoprotein(a), not LDL is the primary risk factor for CVD in human plasma. Lipoprotein(a) is a unique particle essentially composed of a LDL particle and an additional adhesive protein designated apoprotein(a) (apo(a)). The adhesive properties of apo(a) are the cause for the selective retention of lipoprotein(a) in the vascular wall and for the accumulation of lipids and lipoproteins inside the wall (Figure 1).
Lipoprotein(a) is an independent risk factor for CVD. None of the epidemiological studies thus far assessing the plasma risk profile for CVD showed any correlation between lipoprotein(a) levels and total-cholesterol or LDL-cholesterol levels. The most conclusive study that lipoprotein(a), not LDL, is the primary risk factor for CVD was carried out in a genetically defined cohort of LDL-receptor deficient patients.5 This genetic disorder is characterized by significantly elevated plasma LDL levels and was thought to lead almost invariably to premature CVD. Surprisingly, 60% of these LDL-receptor deficient patients had no clinical signs of CVD, while 40% had developed CVD. Both groups did not differ in their extremely high plasma levels of LDL-cholesterol (above 300 mg/dl) or of total cholesterol (390 mg/dl). The two groups differed, however, significantly in their lipoprotein(a) plasma levels and CVD patients had on average three-fold higher plasma lipoprotein(a) levels. This study in a large group of patients selected to minimize genetic variations allows the following conclusions:
Elevated plasma lipoprotein(a) is the primary risk factor for CVD. Increased LDL levels, in addition to elevated lipoprotein(a) levels, increase the risk for CVD. High plasma LDL levels alone are not associated with an increased risk for CVD.

Figure 1.
Equally strong evidence that lipoprotein(a), not LDL, is the primary risk factor for CVD comes from a recent re-evaluation of the Framingham Heart Study, one of the largest prospective epidemiological studies determining the risk profile for CVD. Lipoprotein(a) ranked among the most prevalent risk factors for heart attacks. Moreover, a given quantity of lipoprotein(a) in the blood conferred as much added risk for CVD as does 10 times the quantity of LDL.6 Lipoprotein(a) was discovered 30 years ago.7 The negligent exclusion of this important risk factor from previous epidemiological studies deserves an explanation. It may in part be provided by methodological difficulties as a result of the structural similarity between lipoprotein(a) and LDL. Plasma lipoproteins in most epidemiological studies were determined by means of the “Friedewald Formula”,8 a method that does not allow the differentiation between LDL and lipoprotein(a). The re-evaluation of all large epidemiological risk factor studies has become necessary. The results of these evaluations will further confirm lipoprotein(a) as the primary risk factor for CVD. The evidence that lipoprotein(a), not LDL, is the primary risk factor for CVD is not limited to human plasma.
Lipoprotein(a), not LDL, is the Primary Risk Factor Contributing to Atherosclerotic Plaques
Present concepts of human atherosclerosis assume that LDL is the main vehicle by which cholesterol and other lipids are deposited in the vascular wall. More recently it has been proposed that cellular uptake of oxidized LDL by macrophages and other scavenger cells and subsequent foam cell formation are the decisive mechanisms for development of atherosclerotic plaques.4 According to this concept foam cell formation or the extracellular deposition of LDL would have to play a decisive role in the progression of atherosclerotic lesions. A closer histological look on the in situ situation of human atherosclerotic lesions challenges this concept. The progression of atherosclerotic deposits is paralleled by a structural impairment of the vascular wall and by the accumulation of lipoprotein(a).
Together with my colleagues at Hamburg University I reported the most comprehensive studies differentiating between the deposition of LDL and lipoprotein(a) in human atherosclerosis. ??¹¹ Although these studies are frequently quoted, their significance for the development of human atherosclerosis is still insufficiently understood. These studies and their correct interpretation have significant implications for future therapeutic approaches for CVD. The conclusions of these studies are marshaled here as follows:
Lipoprotein(a) is the predominant risk factor contributing to the progression of atherosclerotic lesions in man.
The amount of lipoprotein(a) deposited in atherosclerotic lesions corresponds with the extent of the lesions.
Lipoprotein(a) is deposited in the extracellular matrix of the vascular wall in the form of largely intact lipoprotein particles, which can be isolated from the wall. This finding implies the reversibility of the lipoprotein(a) deposition in the vascular wall.
Isolated LDL deposition was rarely found and LDL alone, without simultaneous lipoprotein(a) deposition, cannot be considered a primary factor determining the advancement of human atherosclerotic lesions.
The adhesive protein apo(a) is responsible for the selective retention of the lipoprotein(a) particle inside the vascular wall compared to LDL and other lipoproteins.
These results do not exclude the deposition of other potentially atherogenic lipoproteins (LDL, very low-density lipoprotein in VLDL) in addition to and in the same areas lipoprotein(a) accumulated. The discovery of the predominant role of lipoprotein(a) in human atherosclerosis and the discovery of its potential reversibility were decisive preconditions directly leading the way to identify the therapeutic approaches discussed below.
Mechanism Leading to the Extracellular Accumulation of Lipoprotein(a) in the Vascular Wall
The extracellular accumulation of lipoprotein(a) in the vascular wall as the predominant pathomechanism of human atherosclerosis is no coincidence. The frequency of this mechanism today is directly related to its advantage during the evolution of man. After the loss of endogenous ascorbate production in our ancestors lipoprotein(a) became a life-saving feature to counteract fatal blood-loss through the scorbutic vascular wall. While scurvy is essentially unknown today, chronic insufficient dietary ascorbate intake is widespread. The deposition of lipoprotein(a) in the vascular wall stabilizes the wall of the arteries particularly during ascorbate deficiency. With insufficient dietary ascorbate intake over decades this defense mechanism overshoots and CVD develops. 1,2
The lipoprotein(a) particle is an ideal defense molecule. Apo(a), an adhesive molecule,¹² interacts with a variety of cellular and extracellular constituents of the vascular wall including collagen, elastin, fibronectin, and glycosaminoglycanes as well as fibrin/ fibrinogen. The apo(a) macromolecule itself as well as the lipoprotein(a) particle confer stability to the structurally impaired vascular wall.
Moreover, the deposition of lipoprotein(a) in the vascular wall can favor the additional retention of other lipoprotein particles such as LDL and VLDL. Lipoprotein(a) has been shown to bind to lipoproteins containing apoB¹³ and the accumulation of LDL and VLDL in addition to elevated lipoprotein(a) levels but not alone.5
With the extracellular deposition of lipoprotein(a) nature developed a sophisticated and reversible mechanism to render compensatory stability to the vascular wall during times when these walls ware weakened by a deficiency of essential nutrients. The reversible deposition of lipoproteins in the vascular wall is a key to new therapeutic approaches. To optimally exert this defense function the lipoprotein(a) particle would inevitably lead to a loss of its function to confer stability.
In contrast to this mechanism, present hypotheses on human atherogenesis presuppose the degradation of the lipoprotein particles into lipids and amino acids by scavenging cells in the vascular wall.4 The importance of these mechanisms in the development of human atherosclerosis needs to be further evaluated. It is, however, evident that these mechanisms are inferior to the extracellular deposition of lipoprotein(a) with respect to two important features: stability and reversibility. This may explain why neither foam cell formation nor the extracellular deposition of LDL are found to parallel the progression of atherosclerotic lesions.
Irrespective of the pathomechanisms of human atherogenesis they can largely be prevented by maintaining the structural integrity, stability, and elasticity of the vascular wall. On the basis of an improved understanding of human CVD presented in the first part of this paper I will now summarize the most important preventive and therapeutic aims for this disease.
Therapeutic Aim #1: Preserving and Restoring the Integrity and Stability of the Vascular Wall
The impairment of the vascular connective tissue and loss of the endothelial barrier functions are the underlying morphologic changes of any form of CVD. The instability of the vascular wall is a prominent risk factor for human CVD explaining the predominantly localized clinical manifestation of this disease in form of heart attack and stroke.¹’² Preserving and restoring the integrity and stability of the vascular wall is the most important therapeutic aim for prevention and treatment of human CVD. Integrity and stability of connective tissue are critically dependent on an optimum amount and function of collagen and elastin. Ascorbate stimulates the production of collagen and elastin and thereby directly contributes to preserving and restoring the stability and integrity of the vascular wall.14
It therefore comes as no surprise that CVD is essentially unknown in animals producing their own vitamin C at a daily rate of several thousand milligrams. Nor is it a surprise that lipoprotein(a) is primarily found in species that had lost the ability of ascorbate synthesis, a discovery I made in 1987. In humans a growing amount of clinical and epidemiological data support the value of ascorbate in the prevention of CVD. A recent epidemiological study in 11,000 Americans showed that dietary ascorbate intake between 200 mg and 500 mg correlated with a reduction in CVD up to 50% and an increase in life expectancy for up to 6 years.15 Beside providing structural stability to the human body, ascorbate is also involved in a variety of enzymatic and other metabolic functions, some of which will be discussed below.
Therapeutic Aim #2: Lowering Lipoprotein(a) Levels in Plasma
Lowering the plasma levels of lipoprotein(a) is the second most important therapeutic aim. Lipoprotein(a) is produced in the liver and the production rate of apo(a) largely determines the plasma levels of this lipoprotein. None of the currently available cholesterol-lowering drugs is known to significantly affect plasma lipoprotein(a) levels. In contrast, optimum dosages of two vitamins, niacin (vitamin B3) and ascorbate have been reported to lower lipoprotein(a) plasma levels.16-18 Their therapeutic mechanism, however, has not yet been explained. I have obtained preliminary in vitro evidence that lipoprotein(a) production can be lowered by increasing the concentration of NADPH. NADPH is involved in a multitude of metabolic regulatory processes. Niacin is a constituent of the NADP molecule and ascorbate can reduce or “re-charge” the NADP molecule to NADPH. Thus ascorbate and niacin could decrease lipoprotein plasma levels – at least in part – by increasing NADPH concentrations (Figure 2).
Beside the lowering of lipoprotein(a) in plasma the risk for CVD can be further reduced by preventing accumulation of this risk factor in the vascular wall.
Therapeutic Aim #3: Preventing the Accumulation of Lipoprotein(a) in the Vascular Wall
Prevention of the accumulation of lipoprotein(a) in the vascular wall is an important therapeutic aim in reducing the risk of CVD. As discussed above the lipoprotein(a) particle can interact with a variety of constituents of the vascular wall. The extracellular deposition of lipoprotein(a) particles in the vascular wall via the adhesive protein apo(a) immediately suggests novel therapeutic approaches. Interfering with the binding of lipoprotein(a) to constituents of the vascular wall will decrease the tendency of this atherogenic lipoprotein to accumulate in the vascular wall and thereby reduce the risk for the development of atherosclerotic lesions.

Figure 2.
The amino acids L-lysine, L-proline, and hydroxyproline can interfere with the binding of lipoprotein(a) to important constituents of the vascular wall.13’1? The use of L-lysine and L-proline to prevent the deposition of atherogenic lipoproteins in the vascular wall opens novel therapeutic avenues. Supplementation of hydroxyproline and hydroxylysine can be rendered redundant by co-administration of ascorbate which can hydroxylate lysine and proline residues.2
L-Lysine
The essential amino acid L-lysine competitively inhibits the binding of lipoprotein(a) to fibrinogen, fibrin, and fibrin degradation products which are known to be hallmarks of the atherosclerotic lesion. My earlier findings about the potential reversibility of lipoprotein(a) deposition and the isolation of lipoprotein(a) by use of lysine led to the therapeutic introduction of L-lysine and lysine analogs in an earlier paper1 (Figure 3a).
L-proline and hydroxyproline
Trieu et al. Reported that lipoprotein(a) also binds to L-proline and hydroxyproline with an even higher affinity than to lysine.13 Since collagen and elastin are particularly rich in proline residues this mechanism is of importance for the binding and retention of the lipoprotein(a) particle in the vascular wall. On the basis of these observations I propose here the therapeutic use of L-proline in the prevention and treatment of CVD. The dietary supplementation of this amino acid should prevent the binding of lipoprotein(a) to collagen and other proline-rich constituents of the vascular wall and thereby prevent the accumulation of lipoprotein(a) in the vascular wall (Figure 3b).
Therapeutic Aim #4: Reversal of Existing Atherosclerotic Lesions by Releasing Lipoprotein(a) from the Vascular Wall
The improved understanding about human atherosclerosis and in particular about the role of lipoprotein(a) discussed in this paper opens the way to a break-through in the treatment of CVD: the pharmaceutical reversal of existing atherosclerotic lesions. The key to this breakthrough is the reversibility of the accumulation of lipoprotein(a) in the vascular wall. Through the same mechanism by which L-lysine and L-proline can prevent lipoprotein(a) deposition, optimum concentrations of these amino acids can release accumulated lipoprotein(a) from the vascular wall. The release of lipoprotein(a) from the atherosclerotic lesions must lead to a reduction of these atherosclerotic deposits and thereby to a reversal of existing CVD.
Dietary supplementation of optimum amounts of L-lysine and L-proline could contribute to releasing lipoprotein(a) deposited in the vascular wall. The experimental evidence for these novel therapeutic options is already available. Comprehensive clinical confirmation should soon lead to the reduction of existing atherosclerotic deposits in CVD patients on the basis of selected nutritional supplements.
Therapeutic Aim #5: Reducing the Risk for CVD from Other Lipids and Lipoproteins
LDL
While the CVD risk for elevated LDL levels alone has to be re-evaluated, elevated LDL levels in addition to elevated lipoprotein(a) levels are known to increase the risk for CVD exponentially.5 This fact can be explained by the following mechanism. LDL can bind to lipoprotein(a) via proline residues (Figure 3c). This binding of LDL to lipoprotein(a) already deposited in the vascular wall can accelerate the development of atherosclerotic lesions.

Figure 3a.

Figure 3b.
In the light of this mechanism, lowering elevated plasma levels of LDL remains a therapeutic aim. In numerous studies niacin as well as ascorbate have been shown to reduce elevated plasma levels of LDL. As with lipoprotein(a) NADPH may play a regulatory role on the synthesis rate of VLDL the precursor of LDL. Moreover, dietary supplementation of L-proline could prevent the binding of LDL to lipoprotein(a) already deposited in the vascular wall and, by the same mechanism, release already deposited LDL from the atherosclerotic lesions.
VLDL
VLDL is a potentially atherogenic precursor of LDL particularly enriched in triglycerides. Niacin and ascorbate have also been shown to be of particular value in lowering VLDL plasma levels. Moreover, optimum L-proline concentrations should also interfere with the binding of VLDL inside the vascular wall.
Thus dietary supplementation of ascorbate and niacin are of particular value to decrease the plasma levels of atherogenic lipoproteins. Optimum dietary supplementation with the amino acids L-lysine and L-proline could release not only lipoprotein(a) but also other atherogenic lipoproteins from the vascular wall.
VLDL and other triglyceride-rich lipoproteins, however, can contribute to atherogenesis also by another mechanism. Their enrichment in fatty acids renders them particularly subjectible to oxidative modification and thereby enhances their atherogenicity.

Figure 3c.
Therapeutic Aim #6: Prevention of Damage from Oxygen Free Radicals
Oxygen free radicals are promoters of atherogenesis. They lead to structural impairment and to oxidative modification of lipoproteins as well as other metabolic constituents.²³ Antioxidant nutrients such as ascorbate, tocopherol (vitamin E) and beta carotene (provitamin A) can protect against oxidative damage and against oxidative modification of lipoproteins. Elevated plasma concentrations of these nutrients have been shown to be associated with a decreased risk of CVD. 23,24 Nutritional supplements with antioxidative properties, including coenzyme Q 10 and selenium, contribute to maintaining optimum cardiovascular health.
Therapeutic Aim #7: Optimum Cellular Function
Optimum function of endothelial cells, myocardial cells, smooth muscle cells, macrophages and other cell system critically determine optimum cardiovascular health. Optimum metabolic function of these cells depends on the availability of essential cofactors for a multitude of biochemical reactions. Of particular importance are pantothenate, a cofactor for acetyl coenzyme A, carnitine for fatty acid transport, the B vitamins for metabolic energy transfer, ascorbate for enzymatic hydroxylations, and coenzyme Q10 in the respiration chain. Optimum availability of these and other essential nutrients, including certain minerals, not only helps protect the vascular system but also improves cardiac function.24 The reduction of the risk for CVD is, of course, also dependent on other factors, such as exercise, cessation of smoking, and a prudent diet.
Conclusion
Effective reduction of the risk for CVD is a primary goal of the health care system in any industrialized country. In this paper I have presented new therapeutic approaches for this disease. Several of my earlier discoveries turned out to be of particular importance for these recommendations: The prominent role of lipoprotein(a) in human atherosclerotic lesions urged for new therapeutic approaches; the isolation of lipoprotein(a) particles from the vascular wall implied the reversibility of human atherosclerosis; the isolation techniques of lipoprotein(a) via lysine suggested the therapeutic use of this amino acid to induce this reversal. The report of the binding of lipoprotein(a) to proline¹³ suggested the therapeutic use of this amino acid in an analogous way. Most importantly my earlier discovery that lipoprotein(a) is primarily found in species which had lost the ability to synthesize ascorbate triggered a series of publications which may significantly improve our understanding of human CVD.¹’²’²º’²6
Ascorbate and several other nutritional supplements are of particular value including niacin, L-proline and L-lysine as well as natural antioxidants. The therapeutic use of these nutrients may pave the was towards a new therapeutic goal: the pharmaceutical, noninvasive reversal of existing CVD with nutritional supplements.
References
Rath M and Pauling L: Solution of the puzzle of human cardiovascular disease: Its primary cause is ascorbate deficiency, leading to the deposition of lipoprotein(a) and fibrinogen/ fibrin in the vascular wall. Journal of Orthomolecular Medicine 1992; 6: 125-134.
Rath M and Pauling L: A unified theory of human cardiovascular disease leading the way to the abolition of this disease as a cause for human mortality. Journal of Orthomolecular Medicine 1992; 7: 5-15.
Brown MS and Goldstein JL: How LDL receptors influence cholesterol and atherosclerosis. Scientific American 1984; 251: 58-66.
Steinberg D, Parthasarathy S, Carew TE, Khoo JC, and Witzum JL: Beyond cholesterol – modifications of low-density lipoprotein that increase its atherogenicity. New England Journal of Medicine 1989; 320: 915-924.
Seed BM, Hopplichter F, Reaveley D, McCarthy S, Thompson GR, Boerwinkle E, and Uterman G: Relation of serum lipoprotein(a) phenotype to coronary heart disease in patients with familial hypercholesterolemia. New England Journal of Medicine 1990; 322: 1494-1499.
Lawn RM: Lipoprotein(a) in heart disease. Scientific American 1992; 266,6: 54-60.
Berg K: A new serum type system in man – The Lp system. Acta Pathologica 1963; 59: 369-382.
Friedewald WT, Levy RI, Fredrickson DS: Estimation of plasma low-density lipoprotein cholesterol concentration without use of the preparative ultracentrifuge. Clinical Chemistry 1972; 18: 499.
Rath M, Niendorf A, Reblin T, Dietel M, Krebber HJ, and Beisiegel U: Detection and quantification of lipoprotein(a) in the arterial wall of 107 coronary bypass patients. Arteriosclerosis 1989;9: 579-592.
Niendorf A, Rath M, Wolf K, Peters S, Arps H, Beisiegel U, and Dietel M: Morphological detection and quantification of lipoprotein(a) deposition in atheromatous lesions of human aorta and coronary arteries. Virchow’s Archiv. A. Pathol. Anat. 1990; 417: 105-111.
Beisiegel U, Niendorf A, Wolf K, Reblin T, and Rath M: Lipoprotein(a) in the arterial wall. European Heart Journal 1990; 11 Suppl. E: 174-183.
Rath M and Pauling L: Apoprotein(a) is and adhesive protein. Journal of Orthomolecular Medicine 1991; 6: 139-143.
Trieu VN, Zioncheck TF, Lawn RM, and McConathy WJ: Interaction of apolipoprotein(a) with apolipoproteinB-containing lipoproteins. Journal of Biological Chemistry 1991; 266: 5480-5485.
Murad S, Grove D, Lindberg KA, Reynolds G, Sivarajah A, and Pinnell S: Regulation of collagen synthesis by ascorbic acid. Proceedings of the National Academy of Sciences USA 1981; 78: 2879-2882.
Enstrom JE, Kanim LE, Klein M: Vitamin C intake and imortality among a sample of the United States population. Epidemiology 1992; 3: 194-203.
Guraker A, Hoeg JM, Kostner G, Papadopoulos NM, Brewer HB Jr: Levels of lipoprotein Lp(a) decline with neomycin and niacin treatment. Atherosclerosis 1985; 57: 293-301.
Carlson LA, Hamsten A, Asplund A: Pronounced lowering of serum levels of lipoprotein Lp(a) in hyperlipidemic subjects treated with nicotinic acid. Journal of Internal Medicine (England) 1989; 226: 271-276.
Rath M: Lipoprotein(a) reduction by ascorbate. Journal of Orthomolecular Medicine 1992; 7: 81-82.
MacLean JW, Thomlinson JE, Kuang WJ, Eaton DL, Chen EY, Fless GM, Scanu AM, and Lawn RM: c-DNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature 1987; 300: 132-137.
Rath M and Pauling L: Hypothesis: Lipoprotein(a) is a surrogate for ascorbate. Proceedings of the National Academy of Sciences USA 1990; 87: 6204-6207.
Pauling L: Case report: Lysine/ ascorbate-related amelioration of angine pectoris. Journal of Orthomolecular Medicine 1991; 6: 144-146.
Halliwell B and Gutteridge JMC: Free radicals in biology and medicine. Oxford University Press, London New York Toronto 1985.
Gey KF, Brubacher GB, and Staehelin HB: Plasma levels of antioxidant vitamins in relation to ischemic heart disease and cancer. American Journal of Clinical Nutrition. 1987; 45: 1368-1377.
Riemersma RA, Wood DA, MacIntyre CCA, Elton RA, Gey KF, and Oliver MF: Risk of angina pectoris and plasma concentrations of vitamin A, C, and E and carotene. Lancet 1991; 337: 1-5.
Folkers K, Vadhanavikit S, and Mortensen SA: Biochemical rationale and myocardial tissue data on the effective therapy of cardiomyopathy with coenzyme Q10. Proceedings of the National Academy of Sciences USA 1985; 62: 901-904.
Rath M: Solution to the puzzle of human evolution. Journal of Orthomolecular Medicine 1992; 7: 73-80.

{kind=link}